
What We Are Learning from the NIH RUNX1-FPD Clinical Research Study
By David Young, M.D., Ph.D.
At this year's RUNX1 Patient Meeting, I shared an update from the NIH RUNX1-FPD Clinical Research Study. I am grateful to the patients and families who attended and asked thoughtful questions, as this is not an easy topic to discuss.
Blood cancer risk may be the most difficult aspect of RUNX1-FPD for patients and families, but it is also one of the most important areas for us to study carefully and openly discuss.
What the NIH Study Has Taught Us So Far

The Anderson family on a recent NIH Clinical Research Study visit with members of the research team.
The NIH RUNX1-FPD Clinical Research Study (also referred to as a RUNX1 Natural History Study) was designed to help us better understand RUNX1 Familial Platelet Disorder (RUNX1-FPD):
how the condition affects people
how those effects change over time
why symptoms and blood cancer risk can vary from one family to another
how we can improve monitoring and care.
The study has connected with nearly 200 families. Almost 600 people have enrolled in the study, including people with confirmed RUNX1-FPD and family members without the condition.
Both groups are important. Many things “run” through families besides RUNX1-FPD, and studying family members unaffected by RUNX1-FPD helps us to identify which health features are related to RUNX1 and which may simply reflect a family's broader health history.
As part of the study, participants may have blood tests, bone marrow biopsies, and other evaluations. Many participants have returned year after year, which helps us see how things change over time. That long-term information is extremely valuable. A single blood test or bone marrow biopsy gives us a snapshot. Repeated visits help us see patterns.
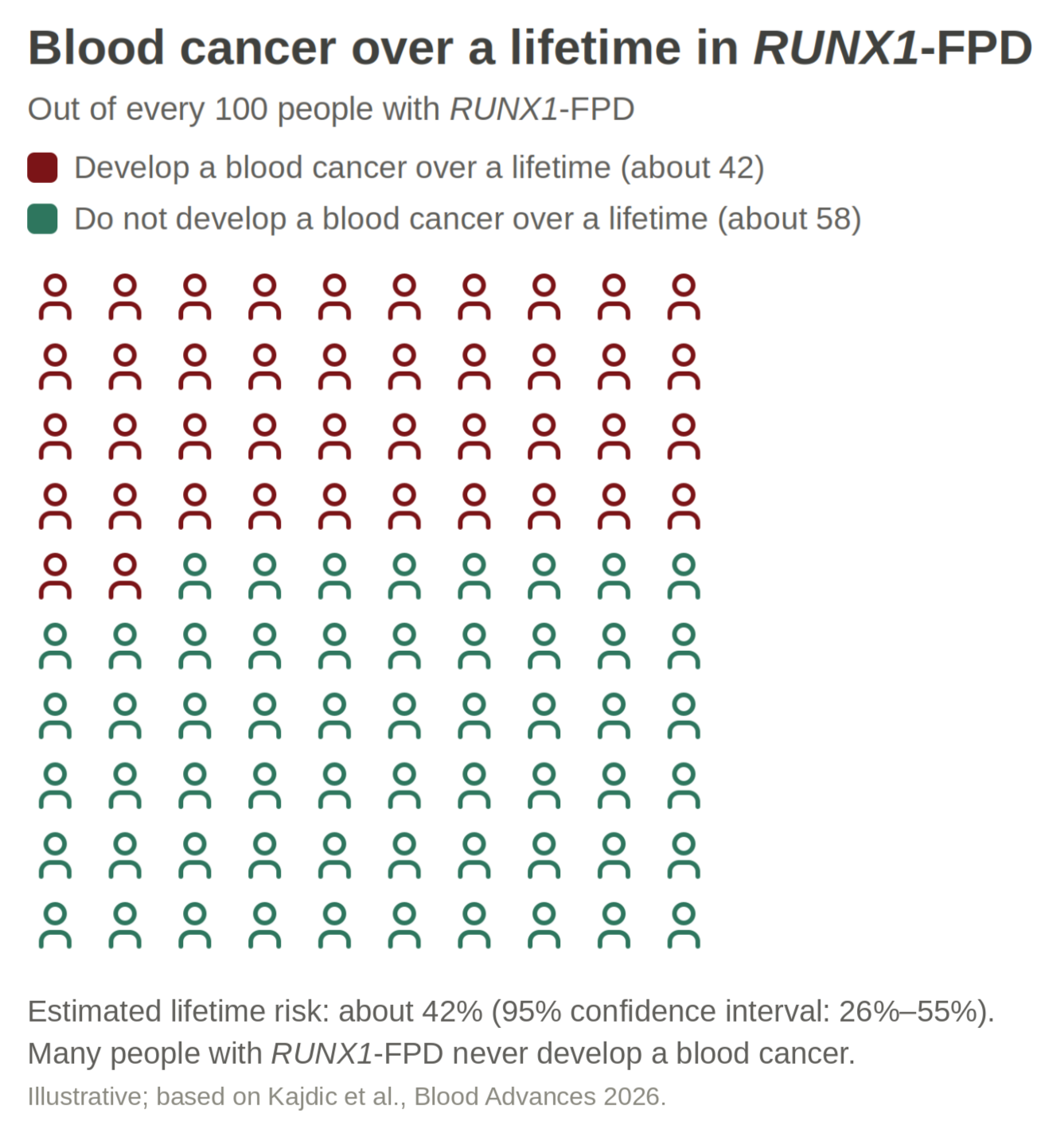
Understanding Blood Cancer Risk in RUNX1-FPD
One of the major questions in RUNX1-FPD is why some people develop blood cancers whereas others do not.
We know that people with RUNX1-FPD have a higher lifetime risk of developing blood cancers. These can include myelodysplastic syndrome, acute myeloid leukemia, and acute lymphoblastic leukemia, but they also include other blood cancers.
Fortunately, our data do not show an increased risk of solid tumors (non-blood cancers) for people with RUNX1-FPD, though we will continue to study this carefully.
Based on the data we have so far, the overall lifetime risk of developing a blood cancer appears to be about 42%, with current estimates ranging from about 26-55%. That number can feel overwhelming, but it is important to remember that risk is not the same as certainty.
Many people with RUNX1-FPD do not develop blood cancer. At the same time, the risk is high enough that careful monitoring is important.
We are also learning that risk can vary from family to family. Some families appear to be more affected by blood cancer, while others appear less affected by it.
Right now, we do not have a simple way to predict a person's blood cancer risk. But it is our goal to one day be able to better understand a person’s risk based on the RUNX1 mutation they carry, their family history, and other factors we hope to identify as we learn more from people and families with RUNX1-FPD.
Pediatric Blood Cancer Risk
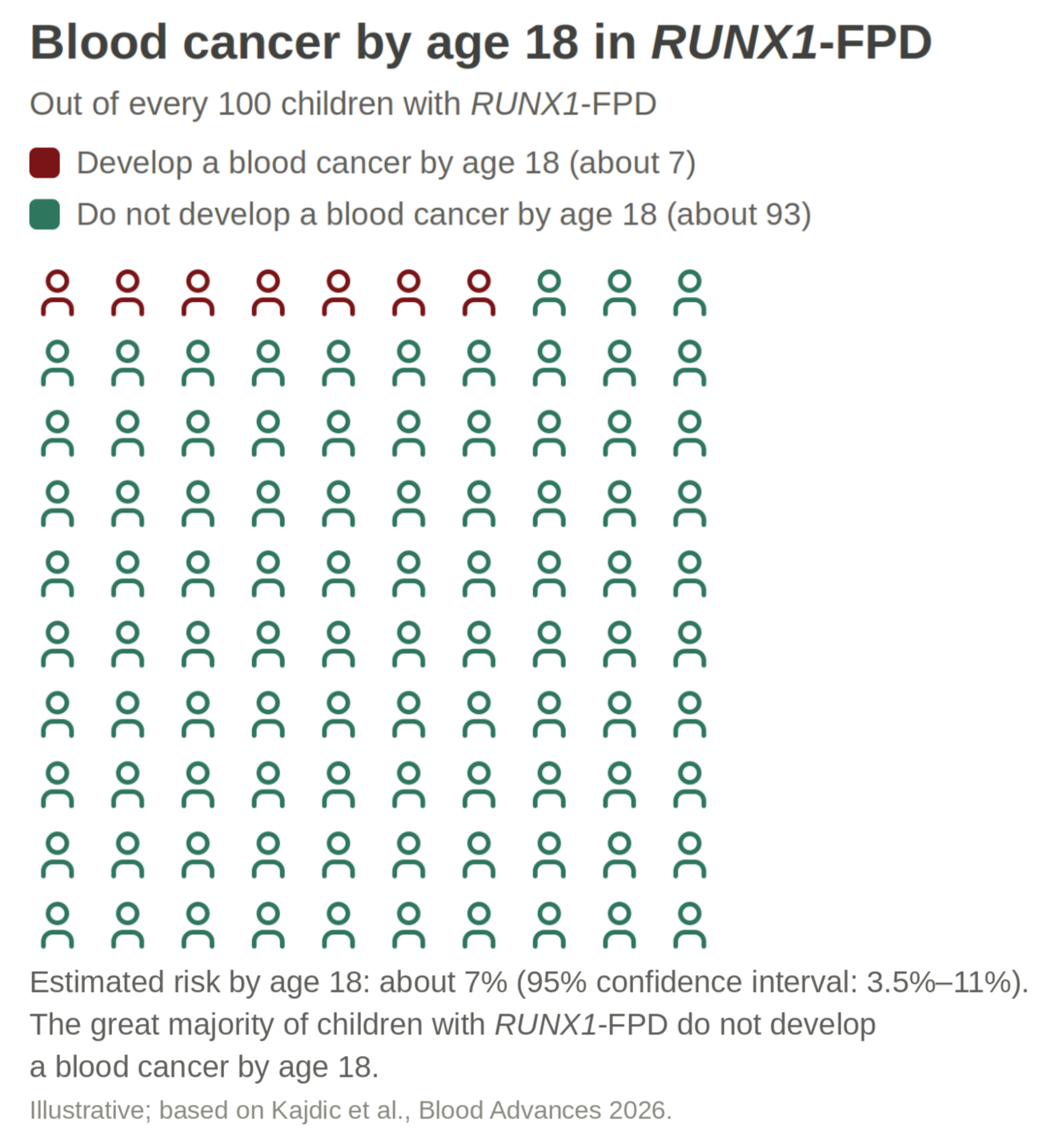
One important new finding we discussed at the Patient Meeting is that blood cancer risk in RUNX1-FPD is not limited to adults.
In the past, many discussions of RUNX1-FPD focused on blood cancer risk in adulthood, especially because the average age of blood cancer diagnosis is in the mid-thirties.
But the NIH data show that risk is not limited to adulthood. There is a period of higher risk in childhood, followed by a more stable period in young adulthood, before risk rises again later.
Between birth and the age of 18, the estimated risk of developing a blood cancer is about 7%. Most children with RUNX1-FPD will not develop cancer in childhood. But this risk is much higher than in the general population (i.e., those without RUNX1-FPD).
This is why we recommend that all relatives in families with RUNX1-FPD be considered for RUNX1 diagnostic testing as soon as possible, including children. We cannot rely only on bruising, bleeding symptoms, or platelet counts to know whether a child has RUNX1-FPD.
Some children may not show obvious signs early on, and genetic testing is the only way to know whether they inherited the family’s RUNX1 mutation. Sadly, some children are only found to have RUNX1-FPD after they first develop a blood cancer.
Knowing whether a child has RUNX1-FPD allows the family and medical team to make informed decisions about care, especially monitoring for cancers, but also taking care of children (and adults) who are at increased risk of unexpected, severe bleeding because of RUNX1-FPD.
What Monitoring May Involve
Monitoring should always be discussed with the patient's own medical team. In general, though, we use several tools to monitor changes over time.
Complete Blood Count with Differential: A CBC with differential is one of the most useful tools we have. This test looks at platelets, red blood cells, and the different white blood cells.
For many patients, a CBC every three to six months may be reasonable. We often suggest every three to four months, especially for children.
Testing more often than that may sometimes create anxiety over small changes that do not mean anything serious, though there are times when additional testing is appropriate.
A CBC may be especially important if someone has:
new, worsening, or unusual bruising or bleeding
increased fatigue
frequent infections
unexplained fevers
weight changes
paleness
abdominal swelling or bloating
When looking at CBC results, trends are often more important than a single number. Blood counts can change for many reasons, including viral infections such as COVID-19 or the flu.
If one result looks different than usual, repeating the test after a few weeks may help show whether the change was temporary or part of a longer-term pattern.
Bone Marrow Biopsies: Bone marrow biopsies are another important monitoring tool. They help doctors look directly at the bone marrow, where blood cells are made. They can also help establish a baseline, so future changes are easier to recognize.
For children, bone marrow biopsies require special consideration, including anesthesia. When there is no specific clinical concern, it may be reasonable to wait until a child is old enough for routine anesthesia, around age 3, before doing a first baseline biopsy. This decision should always be made in partnership between the child’s parents and his or her care team.
For ongoing monitoring, our current recommendation is a yearly bone marrow biopsy with genetic testing of the sample to detect changes over time, though the appropriate schedule should always be discussed with each patient's care team.
We recognize that this is a significant commitment. After several years of stable results, it may become reasonable to space biopsies further apart, especially in late childhood or early adulthood. We are still trying to understand the best long-term approach, and this includes a discussion with an international group of scientists, clinicians, and geneticists to balance the risks and benefits of any particular approach or schedule.
Patients should also feel empowered to discuss comfort and safety with their care team during bone marrow biopsies. Some people do well with local numbing medicine alone. Others may need additional support, such as conscious sedation.
Somatic Mutations and What They May Mean
Another area of active research is understanding which somatic mutations may contribute to blood cancer development. These are genetic changes acquired over time in blood cells. They are not inherited from a parent and are not passed down to children. Instead, they happen after birth, during a person's life.
Some may contribute to cancer development, while others may not. Everyone develops some of these changes as they get older. However, in RUNX1-FPD, we often see them earlier and more often than we would expect in the general population. Some of the genes in which researchers have most often observed these acquired changes in adults include BCOR and TET2.
Finding one or more of these changes does not necessarily mean that a person has cancer or will develop cancer. For example, BCOR changes are relatively common in people with RUNX1-FPD. On its own, having BCOR mutations does not appear to be a warning sign.
What may matter most is the pattern over time. Is the proportion of cells in the bone marrow with this mutation growing? Are new changes appearing? Are blood counts changing, too? These are the kinds of questions that long-term monitoring may help answer. In adults, especially, testing for these mutations may help us detect warning signs earlier.
In children, the picture appears to be different. Children who develop blood cancers are less likely than adults to show these somatic mutation warning signs beforehand. Instead, other kinds of genetic changes, such as changes to whole chromosomes, appear to play a larger role.
That means we cannot rely on genetic testing alone, especially in children. There are other tests used to detect chromosomal changes. It is one of the reasons that blood tests, bone marrow evaluations, and long-term follow-up all remain important.

We have learned a great deal, but many areas of study remain.
These unanswered questions are why we so greatly appreciate the efforts, big and small, by everyone who has participated in the NIH RUNX1-FPD Clinical Research Study. This includes all of the scientists, clinicians, and collaborators who have contributed to this work.
But most importantly, this is only possible thanks to all of the individuals with RUNX1-FPD and their family members who have contributed their time and efforts to the study.
With continued partnership between patients, families, clinicians, researchers, and the RUNX1 community, we are learning more every year.
The findings shared here were published in Hematologic malignancies in pediatric patients with RUNX1-familial platelet disorder with associated myeloid malignancy. Kajdic A, Deuitch NT, Bresciani E, et al. Blood Adv. 2026.
This research was supported by the Intramural Research Program of the National Human Genome Research Institute (NHGRI) in collaboration with the National Cancer Institute (NCI) and National Heart, Lung, and Blood Institute (NHLBI); all of the National Institutes of Health (NIH). The contributions of the NIH authors are considered Works of the United States Government. The findings and conclusions presented are those of the authors and do not necessarily reflect the views of the NIH or the U.S. Department of Health and Human Services.
For more information, please contact either Dr. David Young or NIH Genetic Counselor Natalie Deuitch.